Research in the Lewis group focuses on identifying solutions to challenging synthetic problems through the development of new catalysts for a variety of key chemical transformations. Small molecule transition metal catalysts, enzymes, and artificial metalloenzymes are being explored toward this end and comprise the three major areas of emphasis within the group.
Developing these new functional materials requires a dynamic and highly interdisciplinary research environment. There are opportunities for rigorous training in organic and organometallic synthesis, protein engineering and evolution, molecular biology, structural and biophysical characterization of proteins, and computational modeling. This experience has uniquely prepared students and postdocs for careers in academia, the chemical, pharmaceutical and biotechnology industries, education, and a variety of other fields. Students are encouraged to exploit all of these tools to develop new catalysts for fundamentally important chemical transformations.
Please see below for a general overview of our research, and check out our Prezi for more details about these projects and the people who make them happen!
Protein Engineering and Directed Evolution
Enzymes are remarkable catalysts that have the potential to greatly improve the efficiency of chemical synthesis. They exhibit extremely high catalytic proficiency and selectivity toward their native substrates, and they typically operate at ambient temperature and pressure within narrow pH and redox potential ranges in aqueous solution. New enzymes are constantly being identified from studies on natural product biosynthesis, genome mining, de novo design, and other approaches. Unfortunately, however, the enzymes obtained from these efforts rarely possess useful levels of activity or selectivity on substrates of interest, and their activity is often compromised under preparative reaction conditions (high concentration, organic co-solvents, etc.). Central to any effort to employ enzymes for chemical synthesis is a robust method to improve their catalytic properties. In the Lewis group, we use directed evolution for this purpose:
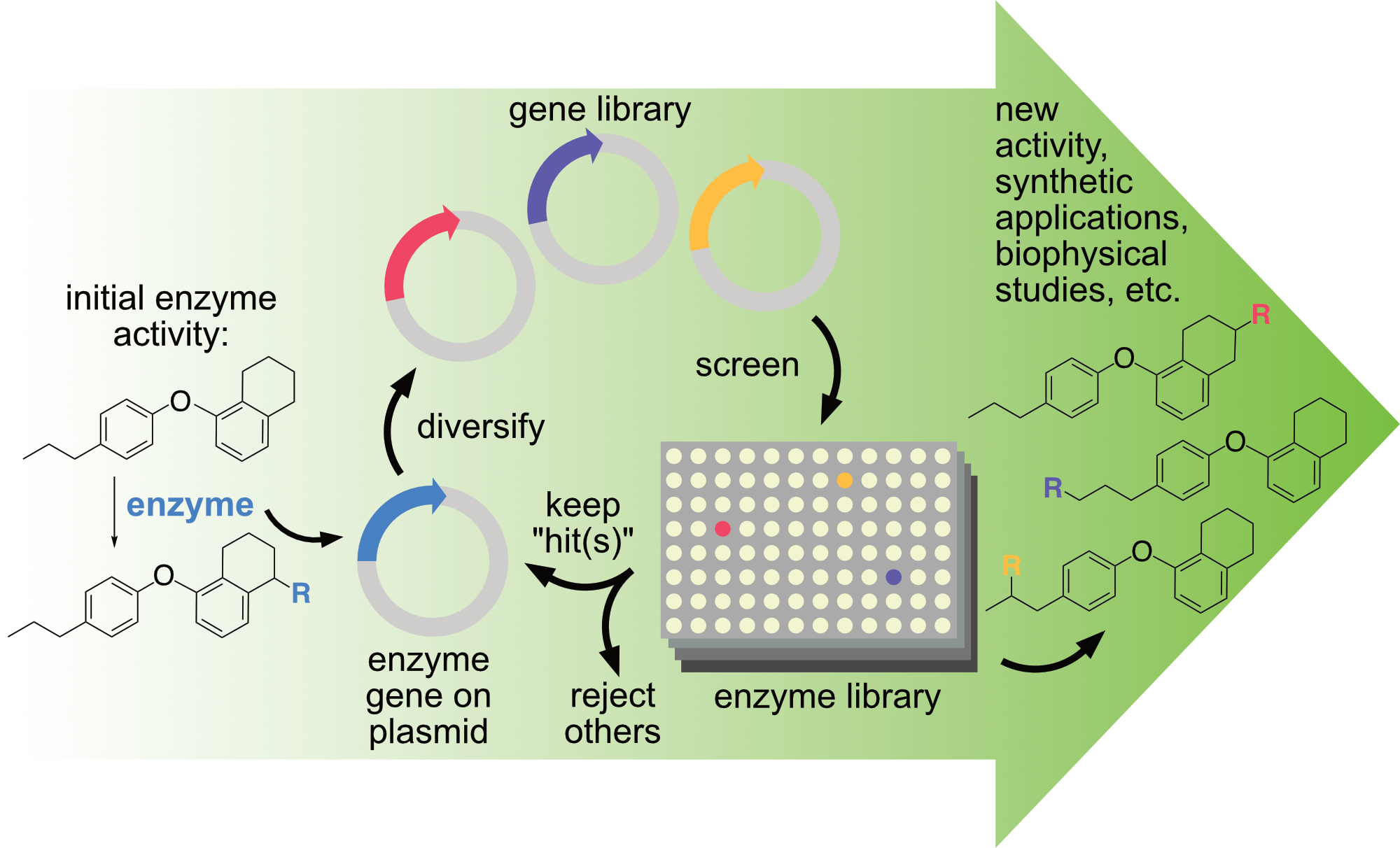
At its core, directed evolution involves diversifying a gene encoding an enzyme of interest, screening the activity of the enzyme variants, and repeating the process on the gene(s) encoding improved variants while rejecting others. In this way, the native activity and selectivity of an enzyme of interest can be rapidly altered or improved to enable new reactions under conditions suitable for preparative reactions. This approach lies at the heart of nearly all projects in the Lewis group. Whether students are working with natural enzymes or artificial metalloenzymes, they have the opportunity to learn state-of-the-art techniques in directed enzyme evolution. Most projects also leverage an integrated robotic platform to automate nearly every step of directed evolution. This system was constructed using funds from a Defense University Research Instrumentation Program (DURIP) award from the U.S. Army Research Office (66796-LS-RIP). It is compatible with enzymes that catalyze a wide range of reactions and provides facile access to library sizes of consisting of thousands variants. See the system in action here:
We are also developing new approaches to construct and screen libraries, including using techniques such as continuous evolution and fluorescence activated cell sorting. While these approaches are more limited in the range of compounds and reactions to which they can be applied, they have the potential to dramatically accelerate the evolution process and thus allow us to probe different questions regarding enzyme evolution and catalysis.
As noted above, directed evolution of proteins involves iterative mutation of a protein and functional screening/selection of the resulting variants to create protein variants with improved properties. Prior to the development of directed evolution, protein engineering, such as it was, largely involved targeted point mutation of proteins and thus only modest improvements in function. Directed evolution revolutionized this process and has led to stunning improvements in protein function for fine/commodity chemical synthesis, selective catalysis, pharmaceuticals development, materials science, chemical biology, and a wide range of other fields. It is safe to say that any application that uses a protein could benefit from some form of directed evolution at some point in its development. Moreover, directed evolution has influenced the means by which protein function and natural evolution is understood and studied. Two seminal and complementary methods, one by Prof. Frances Arnold published in 1993 and one by Dr. Willemm (Pim) Stemmer (deceased) published in 1994, mark the advent of directed evolution, and the pair received the 2011 Draper Prize for their discoveries. One half of the 2018 Nobel Prize in Chemistry was awarded to Prof. Arnold for her ground-breaking work on “the directed evolution of enzymes”. Prof. Lewis was a postdoctoral fellow with Prof. Arnold at Caltech from 2008-2010.
Biocatalysis
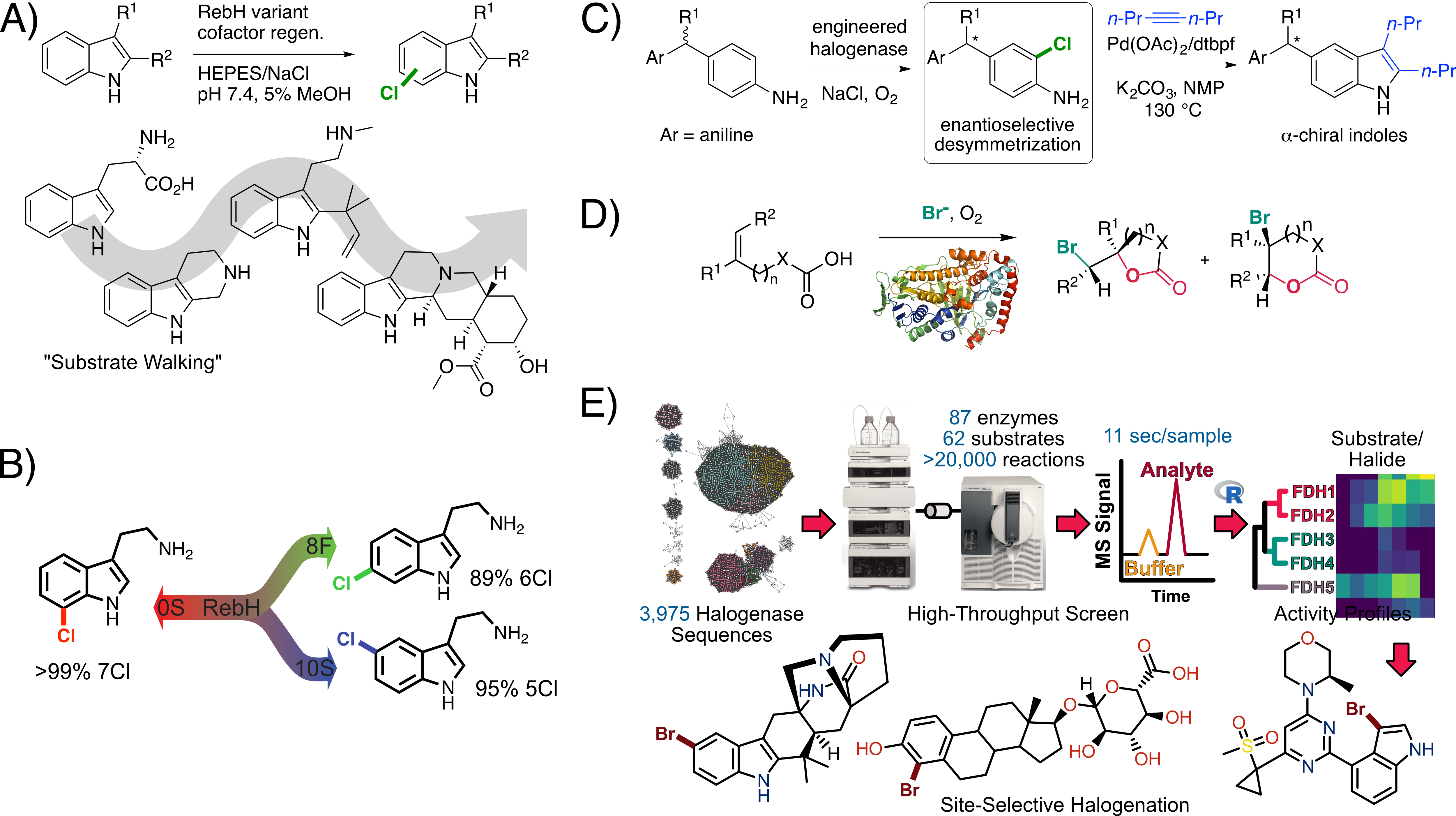
We are exploiting this property to engineer various flavin dependent halogenases (FDHs) for use in organic synthesis due to the importance of halogenated compounds as both building blocks and active pharmaceutical ingredients. Our group has reported the first examples in which directed evolution of FDHs (typically the tryptophan halogenase RebH) has been used to improve stability, expand substrate scope (Fig. 1A), achieve high selectivity for different sites on a given substrate (Fig. 1B), and enable enantioselective (Fig. 1C)and atroposelective (in progress) transformations. Recently, we also found that evolved FDHs can catalyze non-native enantioselective halocyclization (Fig. 1D), providing access to halogenated aliphatic compounds rather than the aromatic compounds generated via native FDH catalysis. We have used genome mining (Fig. 1E) to identify FDHs that provide unique function relative to our evolved enzymes, further expanding the utility of FDHs for chemical synthesis. Finally, we explore structure function relationships in improved enzymes to help rationalize mechanisms for the observed improvements and to inform subsequent engineering efforts. More recently, we have been exploring polymer degradation by lipases and non-native enzyme catalysis by B12-dependent proteins and enzymes and Fe(II)- and α-ketoglutarate-dependent oxygenases and halogenases (in progress).
Fig. 1. A-D) Examples of FDH directed evolution. E) Genome mining to identify new FDHs for biocatalysis.
Artificial Metalloenzymes
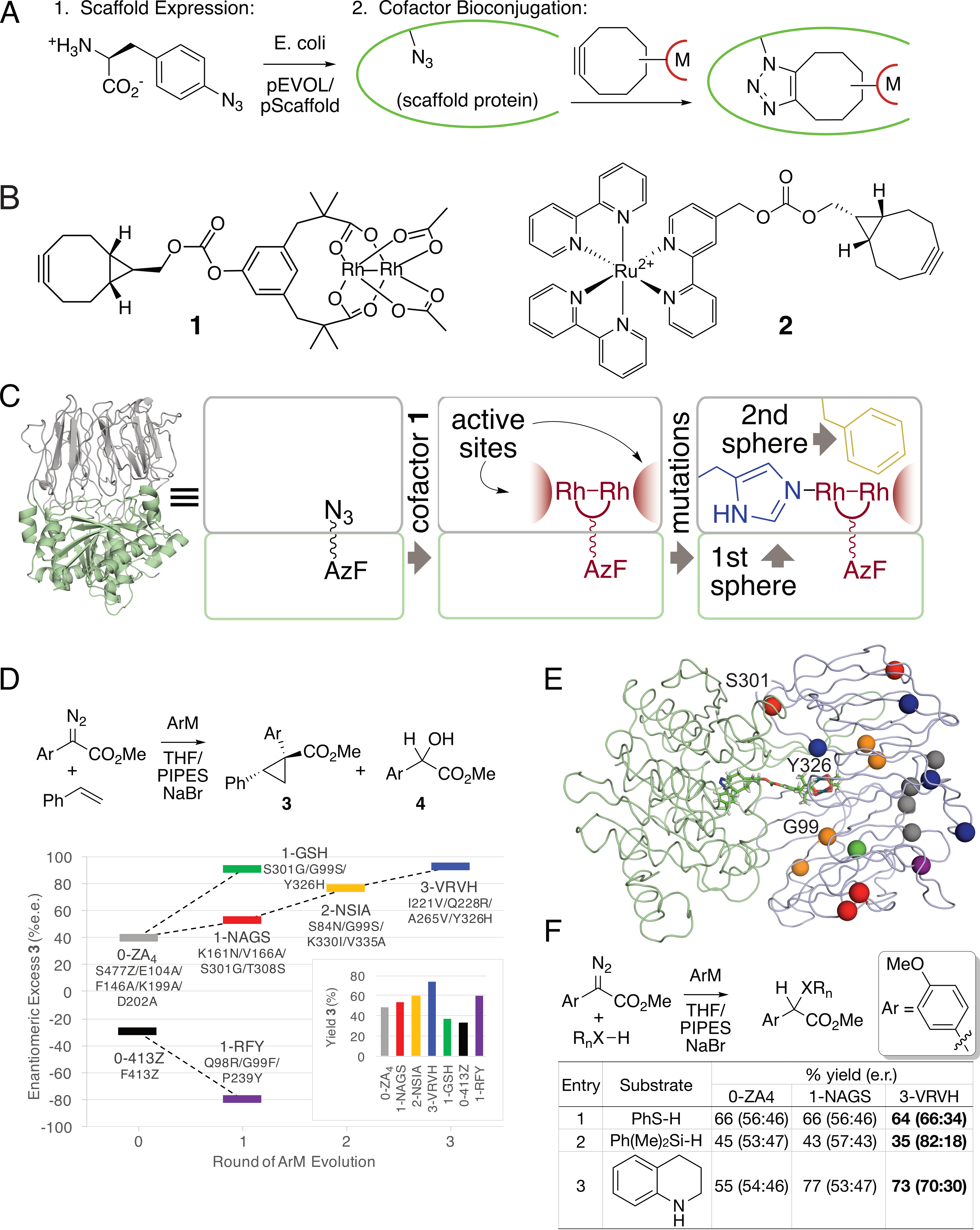
Many powerful reactions, particularly those catalyzed by non-biological metals, are not found in nature, so systems that combine the reactivity of metal catalysts with the evolvability and specificity of enzymes are highly sought after. To expand the scope of reactions are developing new classes of artificial metalloenzymes (ArMs), hybrid constructs comprised of protein scaffolds and metal catalysts. Our group pioneered the use of strain-promoted azide alkyne addition using genetically-encoded azide residues to facilitate ArM formation (Fig. 2A) using a variety of cofactors (Fig. 2B). We have subsequently focused on ArMs generated by linking these cofactors to a prolyl oligopeptidase (POP) from Pyrococcus furious. For example, optimization of dirhodium ArMs generated from cofactor 1 using both rational design (Fig. 2C) and directed evolution (Fig. 2D/E) is being used to produce highly active enzymes for a variety of carbene addition and insertion reactions (Fig. 2F). We recently showed that directed evolution can also be used to engineer dirhodium ArMs to enable a cascade process with an ene reductase that otherwise requires intermediate isolation. Very recently we showed that we could incorporate ruthenium cofactor 2 into POP to modulate its photophysical properties, and, in a related effort, use genetically encoded bipyridine residues to control the conformation and native peptidase catalysis of POP.
Fig. 2. A) ArM formation via strain-promoted azide-alkyne cycloaddition. B) representative dirhodium and ruthenium cofactors. C) General scheme for ArM formation within a prolyl oligopeptidase from pyrococcus furiosus. D) Improved selectivity and yield for cyclopropanation catalyzed by ArMs generated via random mutagenesis. E) Location of mutations in improved ArM variants. F) Improved yield and selectivity for other carbene insertion reactions using evolved ArMs.
In parallel with our ArM design and evolution efforts, we are pursuing detailed mechanistic studies on the systems developed in our laboratory to date. These studies involve steady state kinetics, biophysical experiments (e.g. FRET, LC-MS/MS, NMR, UV/Vis, etc.), and computational simulation (MD, QM/MM in collaboration with the Roux group at Chicago). Beyond shedding light on the function of these complex and fascinating systems, these studies are intended to inform future design efforts aimed at improving ArM activity and selectivity for synthetic applications.
Transition Metal Catalysis
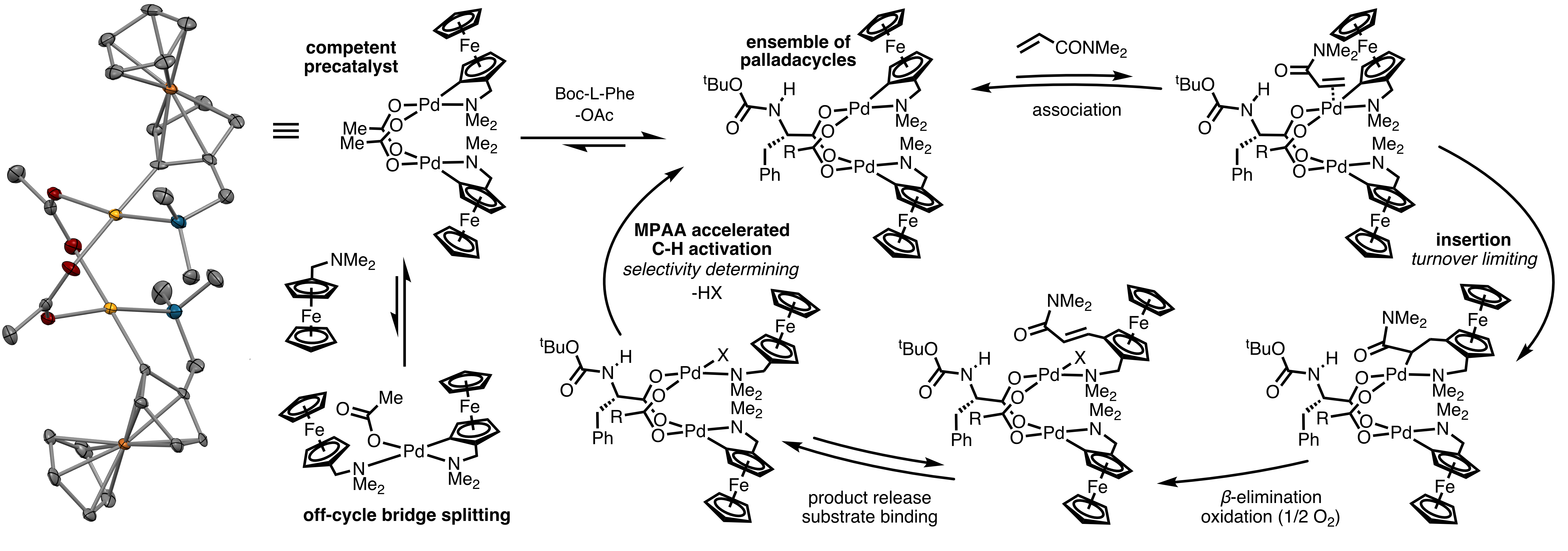
Historically, bioinorganic chemists have sought to synthesize coordination complexes that mimic the structures, spectroscopic features, and even functions of metalloenzyme cofactors that must otherwise be studied, often with great difficulty, in the context of their host enzymes. A key lesson from these efforts is that enzymes play enormous roles in modulating the structures and functions of their metallocofactors. This lesson suggests that ArMs generated by incorporating well-characterized, stable catalysts into protein scaffolds (as outlined above) represent only a subset of the structures that could be possible. For example, in a reversal of the classical bioinorganic chemistry paradigm, proteins could be used to stabilize structures that cannot be readily accessed in solution or maintained throughout a catalytic cycle. Mechanistic studies on small molecule transition metal catalysts could inform the design of protein scaffolds that provide access to putative intermediates to both verify their catalytic relevance and improve their catalytic properties. Our first forays into this area are focused on mono- and dipalladium complexes inspired by our finding that many reactions catalyzed by mixtures of Pd(II) salts and mono-protected amino acid (MPAA) ligands are actually catalyzed by MPAA-bridged Pd(II) dimers. These efforts are in their infancy but progressing rapidly, so please check back to see how this project evolves.